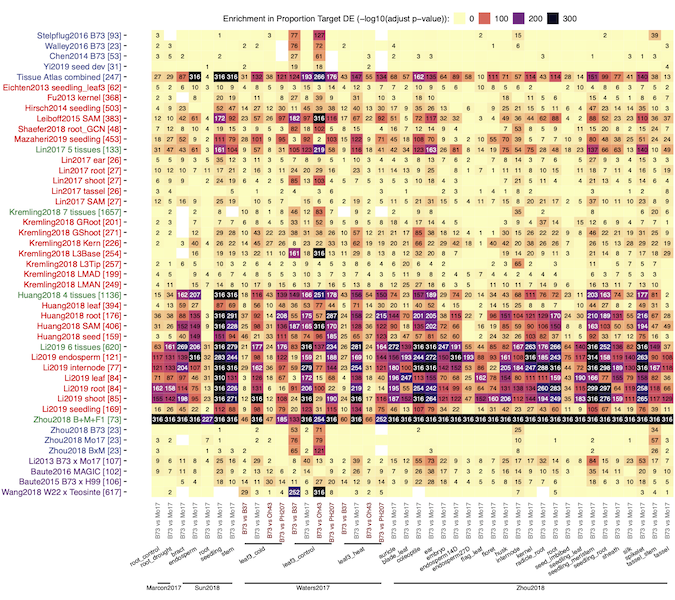
Regulation of gene expression is central to many biological processes. Gene regulatory networks (GRNs) link transcription factors (TFs) to their target genes and represent a map of potential transcriptional regulation. A consistent analysis of a large number of public maize transcriptome datasets including >6000 RNA-Seq samples was used to generate 45 co-expression based GRNs that represent potential regulatory relationships between TFs and other genes in different populations of samples (cross-tissue, cross-genotype, tissue-and-genotype, etc). While these networks are all enriched for biologically relevant interactions, different networks capture distinct TF-target associations and biological processes. By examining the power of our co-expression based GRNs to accurately predict co-varying TF-target relationships in natural variation datasets we found that presence/absence expression changes - rather than quantitative changes - of a TF, are more likely to associate with target gene changes. Integrating information from our TF-target predictions and previous eQTL mapping results provided support for 68 TFs underlying 74 previously identified trans-eQTL hotspots spanning span a variety of metabolic pathways. This study highlights the utility of developing multiple GRNs within a species for detecting putative regulators of important plant pathways and providing potential targets for breeding or biotechnology applications.
Meta Gene Regulatory Networks in Maize Highlight Functionally Relevant Regulatory Interactions
Zhou P, Li Z, Magnusson E, Gomez Cano FA, Crisp PA, Noshay J, Grotewold E, Hirsch C, Briggs SP, Springer NM
2020.
Plant Cell