Highlights:
-
The availability of an increasing number of plant genomes and the development of new high-throughput approaches is resulting in the identification of large collections of transcription factor-target gene interactions.
-
Natural variation is being utilized in a number of important crops to conduct genome-wide association studies to link DNA-sequence variation or changes in gene expression with phenotypes, often quantitative.
-
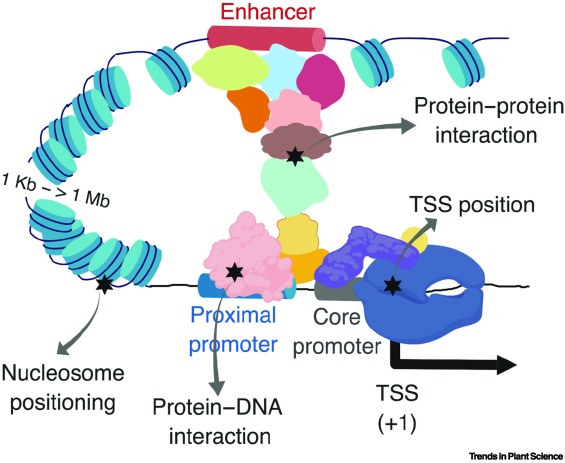
Many trait-associated DNA variants are located in gene regulatory regions, suggesting significant potential in linking gene regulatory information with breeding for complex phenotypes.
-
Fine-tuning the regulation of transcription factors could generate novel variation that would influence plant traits.
Improvement of agricultural species has exploited the genetic variation responsible for complex quantitative traits. Much of the functional variation is regulatory, in cis-regulatory elements and trans-acting factors that ultimately contribute to gene expression differences. However, the identification of gene regulatory network components that, when modulated, will increase plant productivity or resilience, is challenging, yet essential to provide increased predictive power for genome engineering approaches that are likely to benefit useful traits. Here, we discuss the opportunities and limitations of using data obtained from gene coexpression, transcription factor binding, and genome-wide association mapping analyses to predict regulatory interactions that impact crop improvement. It is apparent that a combination of information from these data types is necessary for the reliable identification and utilization of important regulatory interactions that underlie complex agronomic traits.