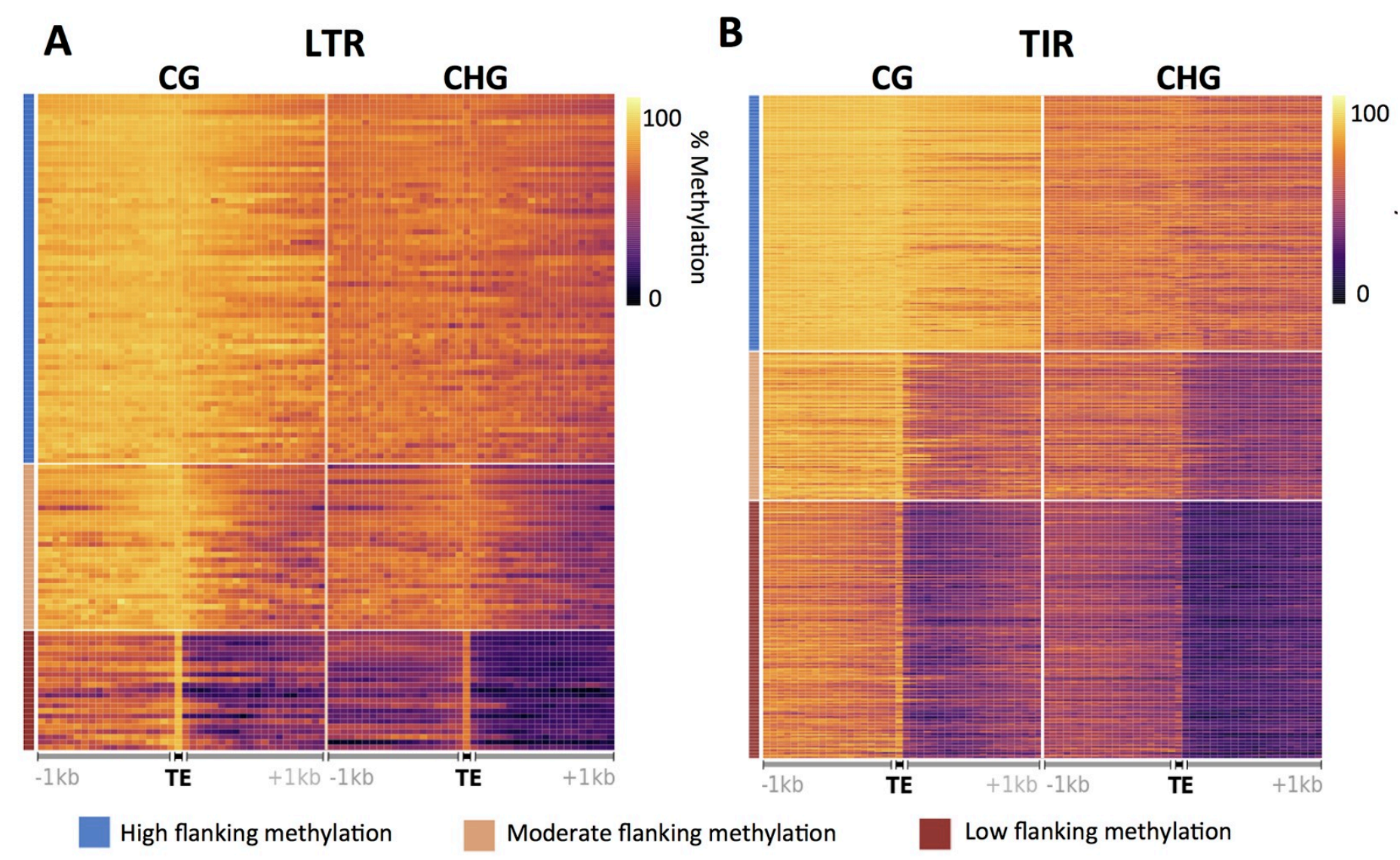
DNA methylation and epigenetic silencing play important roles in the regulation of transposable elements (TEs) in many eukaryotic genomes. A majority of the maize genome is derived from TEs that can be classified into different orders and families based on their mechanism of transposition and sequence similarity, respectively. TEs themselves are highly methylated and it can be tempting to view them as a single uniform group. However, the analysis of DNA methylation profiles in flanking regions provides evidence for distinct groups of chromatin properties at different TE families. These differences among TE families are reproducible in different tissues and different inbred lines. TE families with varying levels of DNA methylation in flanking regions also show distinct patterns of chromatin accessibility and modifications within the TEs. The differences in the patterns of DNA methylation flanking TE families arise from a combination of non-random insertion preferences of TE families, changes in DNA methylation triggered by the insertion of the TE and subsequent selection pressure. A set of nearly 70,000 TE polymorphisms among four assembled maize genomes were used to monitor the level of DNA methylation at haplotypes with and without the TE insertions. In many cases, TE families with high levels of DNA methylation in flanking sequence are enriched for insertions into highly methylated regions. The majority of the >2,500 TE insertions into unmethylated regions result in changes in DNA methylation in haplotypes with the TE, suggesting the widespread potential for TE insertions to condition altered methylation in conserved regions of the genome. This study highlights the interplay between TEs and the methylome of a major crop species.
Monitoring the interplay between transposable element families and DNA methylation in maize
Noshay JM, Anderson SN, Zhou P, Ji L, Ricci W, Lu Z, Stitzer MC, Crisp PA, Hirsch CN, Zhang X, Schmitz RJ, Springer NM
2019.
PLoS Genetics