Abstract
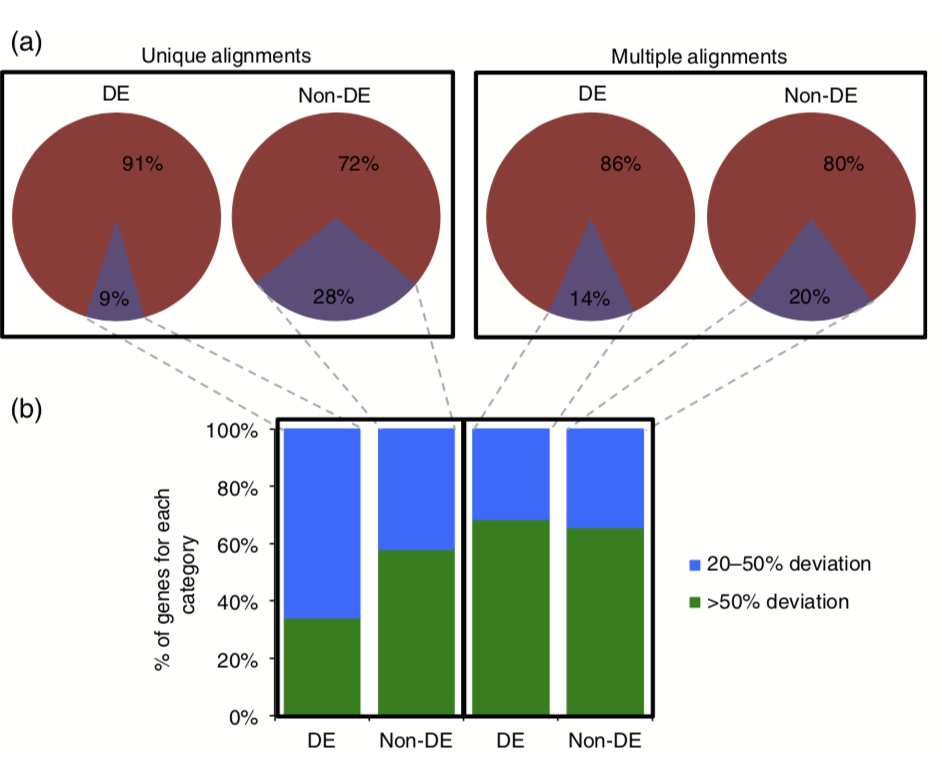
The field of genomics has grown rapidly with the advent of massively parallel sequencing technologies, allowing for novel biological insights with regards to genomic, transcriptomic, and epigenomic variation. One widely utilized application of high-throughput sequencing is transcriptional profiling using RNA sequencing (RNAseq). Understanding the limitations of a technology is critical for accurate biological interpretations, and clear interpretation of RNAseq data can be difficult in species with complex genomes. To understand the limitations of accurate profiling of expression levels we simulated RNAseq reads from annotated gene models in several plant species including Arabidopsis, brachypodium, maize, potato, rice, soybean, and tomato. The simulated reads were aligned using various parameters such as unique versus multiple read alignments. This allowed the identification of genes recalcitrant to RNAseq analyses by having over- and/or under-estimated expression levels. In maize, over 25% of genes deviated by more than 20% from the expected count values, suggesting the need for cautious interpretation of RNAseq data for certain genes. The reasons identified for deviation from expected expression varied between species due to differences in genome structure including, but not limited to, genes encoding short transcripts, overlapping gene models, and gene family size. Utilizing existing empirical datasets we demonstrate the potential for biological misinterpretation resulting from inclusion of ‘flagged genes’ in analyses. While RNAseq is a powerful tool for understanding biology, there are limitations to this technology that need to be understood in order to improve our biological interpretations.